Muchas personas con leucemia mieloide crónica no
presentan síntomas cuando son diagnosticadas con la enfermedad. Con frecuencia
la leucemia se detecta cuando el médico pide exámenes de sangre por algún
problema médico no relacionado o durante un examen físico de rutina.
Si los síntomas y signos sugieren que uno puede tener
leucemia, el médico necesitará analizar muestras de sangre y de médula ósea
para estar seguro de este diagnóstico.
Aspiración y biopsia de la médula ósea
Estas dos pruebas usualmente se hacen al mismo tiempo.
Las muestras se toman de la parte posterior del huso de la pelvis (cadera),
aunque en algunos casos la aspiración se puede tomar del esternón.
En el procedimiento de aspiración de
médula ósea, el paciente se acuesta sobre una mesa (ya sea sobre su costado o
su estómago). Después de limpiar el área, la piel que se encuentra sobre la
cadera y la superficie del hueso se adormece con un anestésico local, que puede
causar una breve sensación de escozor o ardor. Luego se inserta una aguja
delgada y hueca en el hueso, y se usa una jeringa para aspirar una pequeña
cantidad (aproximadamente una cucharada) de médula ósea líquida. Hasta con el
uso de un anestésico, la mayoría de las personas sienten algo de dolor breve
cuando se extrae la médula ósea.
Generalmente se realiza una biopsia de
médula ósea inmediatamente después de la aspiración. Se extrae un pequeño trozo
de hueso y de médula (aproximadamente 1/16 de pulgada de diámetro y 1/2 pulgada
de largo) con una aguja ligeramente más grande que se hace girar al empujarse
en el hueso. La biopsia también puede causar algo de dolor brevemente. Una vez
que se hace la biopsia, se aplica presión en el sitio para ayudar a prevenir el
sangrado.
Estas pruebas se envían al laboratorio, y se analizan
con un microscopio para ver si hay células leucémicas. Estas pruebas también se
pueden hacer después del tratamiento para ver si la leucemia está respondiendo
al mismo.
Pruebas
de laboratorio
Prueba bioquímica de la sangre
Miden la cantidad de ciertas sustancias para detectar
problemas hepáticos o renales causados por la prolongación de la célula de la
leucemia o por los efectos secundarios de ciertos medicamentos
quimioterapéuticos.
Examen de rutina con un microscopio
Un patólogo, un hematólogo y oncólogo observa las
muestras de sangre y médula ósea bajo el microscopio para analizar el tamaño y
la forma de las células en las muestras y determinar si contienen gránulos
(pequeñas manchas que se observan en algunos tipos de glóbulos blancos).
Una característica importante de una muestra de médula
ósea es la porción de ella que consiste células productoras de sangre. Esto se
conoce como celularidad. La médula ósea normal contiene médulas productoras de
sangre y células grasas. Cuando la médula ósea tiene más células productoras de
sangre que lo esperado, se dice que es hipercelular. Si se encuentra muy pocas se llaman hipocelular.
En el caso de leucemia mieloide crónica, la médula ósea a menudo es
hipercelular debido a que está llena de células leucémicas.
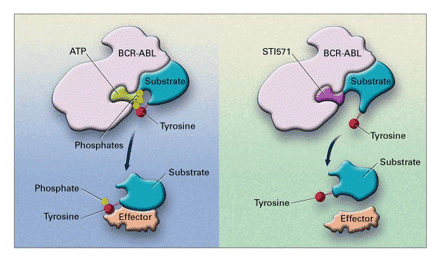
Pruebas genéticas
Esta
prueba consiste en determinar la presencia del cromosoma Filadelfia y/o el gen
BCR-ABL. Este tipo de prueba se usa para confirmar el diagnóstico de CML.
Citogenética
convencional:
Esta
prueba involucra la observación de cromosomas con un microscopio para detectar
cualquier cambio. También se llama determinación del cariotipo. Debido a que
los cromosomas se pueden ver mejor cuando la célula se está dividiendo, una
muestra de sangre o de médula ósea tiene que ser cultivada, de modo que las
células comiencen a dividirse. Esta técnica requiere tiempo y no siempre tiene
éxito.
Las
células humanas normales tienen 23 pares de cromosomas, cada una de las cuales
tiene un tamaño definido. Las células leucémicas de muchos pacientes con CML
contienen un cromosoma anormal llamado cromosoma Filadelfia, que tiene la
apariencia de una versión más corta del
cromosoma 22. Esto sucede cuando se intercambian secciones entre los cromosomas
9 y 22. La detección del cromosoma Filadelfia es útil en el diagnóstico de CML.
Aunque no se puede ver el cromosoma Filadelfia, frecuentemente otras pruebas
pueden detectar el gen BCR-ABL.
Hibridación
in situ con fluorescencia
En
esta prueba se utilizan tintes fluorescentes especiales que sólo se adhieren a
ciertos genes o partes de los cromosomas. En la CML, la prueba FISH se puede
usar para observar secciones específicas del gen BCR-ABL en los cromosomas. En
la CML, la prueba FISH se puede usar para observar secciones específicas del
gen BCR-ABL en los cromosomas. Se puede usar en muestras regulares de sangre o
de médula ósea sin necesidad de primero hacer cultivos de las células. Por lo
tanto, los resultados pueden estar disponibles más rápidamente que con las
citogenéticas convencionales.
Reacción
en cadena de la polimerasa (PCR)
Es
una prueba súper sensible que se puede usar para identificar el oncogén BCR-ABL
en las células de la leucemia. Puede hacerse en muestras de sangre o de médula
ósea y puede detectar cantidades muy pequeñas de BCR-ABL, aun cuando los
médicos no puedan detectar el cromosoma Filadelfia en las células de la médula
ósea mediante pruebas citogenéticas. La PCR se puede usar para ayudar a
diagnosticar CML y también es útil después del tratamiento para ver si aún hay
copias del gen BCR-ABL. Si aún se encuentran copias de este gen, esto significa
que la leucemia sigue presente, aun cuando las células no son detectables con
un microscopio.
Estudios por imágenes
Los estudios por imágenes producen fotos del interior
del cuerpo. Aunque no son necesarios para diagnosticar la CML, pueden servir
para identificar la causa de los síntomas o para ver si el bazo o el hígado
están agrandados.
Tomografía computarizada
Este estudio puede ayudar a detectar si cualquiera de
sus ganglios linfáticos u órganos está agrandado. Generalmente no se necesita
para diagnosticar la CML, pero puede hacerse si su médico sospecha que la
leucemia se está desarrollando en un órgano, como su bazo.
La tomografía computarizada es un tipo de rayos que
produce imágenes transversales detalladas del cuerpo. Antes del estudio, a menudo se le pide que
tome una solución de contraste que ayuda a delinear mejor las áreas normales en
el cuerpo, lo que ayuda a delinearse las áreas anormales más claramente. Esta inyección
puede causar una sensación de sonrojo. Algunas personas son alérgicas al
colorante y desarrollan urticaria. Rara vez presentan reacciones más graves
como problemas para respirar y baja presión arterial.
En algunos casos se puede usar una tomografía
computarizada para guiar con precisión una aguja de biopsia hacia la anomalía
sospechada, como un absceso.
Este procedimiento es llamado biopsia por aguja guiada
por tomografía computarizada, en el cual el radiólogo mueve una aguja de
biopsia a través de la piel y hacia la localización de la masa. Las tomografías
computarizadas se repiten hasta que la aguja está dentro de la masa. Entonces,
se extrae una muestra y se observa con un microscopio. Esto se necesita en muy
pocas ocasiones para la CML.
Imágenes por resonancia magnética
Las imágenes por RM son particularmente útiles para
examinar el cerebro y la médula espinal. Se utiliza ondas de radio e imanes
potentes en lugar de rayos X. la energía de las ondas de radio es absorbida por
el cuerpo y luego liberada en patrón formado por el tipo de tejido del cuero y
por ciertas enfermedades. No solo
proporciona secciones transversales del cuerpo sino también imágenes de
secciones que son paralelas a la longitud del cuerpo. Se puede inyectar material
de contraste pero se hace con menos frecuencia.
Ecografía (ultrasonidos)
En la ecografía o ultrasonidos, se utilizan ondas
sonoras y el eco que estas crean para producir una imagen de los órganos o masa
interna. Con más frecuencia, para esta prueba se coloca sobre su piel un
pequeño instrumento que parece un micrófono y se llama transductor el cual
primero se lubrica con gel. La ecografía emite ondas de sonido y recoge el eco
que rebota desde los órganos. Una computadora convierte el eco en una imagen
que aparece en la pantalla.
Se puede usar para
observar los ganglios linfáticos cercanos a la superficie del cuerpo o para observar
órganos inflamados dentro de su abdomen. Como los riñones, el hígado, y el
bazo.
CML o LMC: leucemia mieloide crónica

.jpg)